Neste artigo, o médico-oftalmologista Dr. Homero Costa, diretor-médico da Ocular Clínica de Olhos, CRM-AL 3702 / RQE 1255, Fellowship em Retina & Vítreo pelo Hospital do Olho (RJ), trata sobre a Retinose Pigmentar, doença degenerativa progressiva da retina e que atinge 1 a cada 4000 pessoas.
RETINOSE PIGMENTAR
A Retinose Pigmentar (RP), também chamada por vários autores de Retinitis Pigmentosa configura-se num dos maiores desafios para Oftalmologia mundial, posto se tratar de uma severa doença neurodegenerativa da retina, numa prevalência de 1 a cada 4000 pessoas, que se inicia com um quadro de “cegueira noturna”, também chamada de nictalopia – o paciente começa a ter muita dificuldade em enxergar em ambientes escuros e/ou com baixa luminosidade -, devido ao extenso dano dos bastonetes (células responsáveis pela visão nestas condições) e que, progressivamente, na maior parte dos casos, evolui para a formação de campo de visão tubular – o paciente enxerga como se tivesse vendo através de um “cano” (figura 1); podendo atingir, nas fases mais tardias, também os cones, que são as células mais especializadas da retina humana, situadas na área central da visão, numa região chamada de mácula e que desempenham a função da visão de cores, além dos detalhes mais finos das imagens, como a nitidez e o brilho destas (figura 2).

Figura 1 – Visão normal (à direita) e com retinose em estágio avançado (à esquerda)
Há, progressivamente, a destruição da retina e de uma camada que fornece suporte nutricional e de proteção para esta, chamada de epitélio pigmentar da retina, levando à formação, nas fases mais tardias de um sinal característico dessa doença que são as “espículas ósseas” (figura 3).

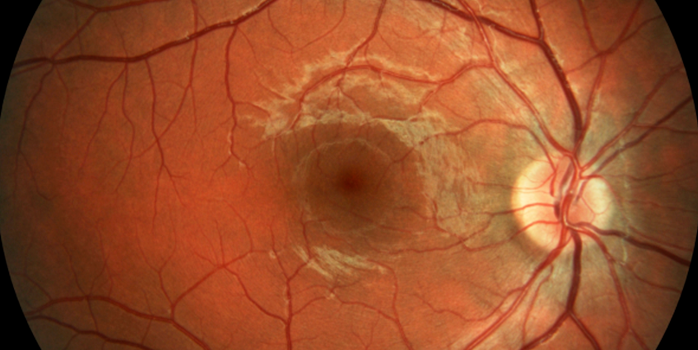
Figura 2 – Retina normal. Área macular (seta azul) Figura 3 – Retinose pigmentar avançada. “Espículas ósseas” (lesões negras) e acometimento macular.
As várias formas de acometimento e gravidade de cada caso dependem da idade de início, a taxa de progressão, a quantidade e a extensão da perda visual são frequentemente relacionadas ao modo de herança genética - existindo em torno de 40 genes ligados às formas dominantes e recessivas desta doença -, mas os casos esporádicos são o grupo mais comum, além de termos aqueles associados a algumas doenças sistêmicas.
Muito já se evoluiu na Oftalmologia moderna e vários tipos de tratamento já foram propostos para tentar frear e até mesmo reverter esta entidade tão avassaladora e debilitante do ponto de vista visual, que é a RP, tais como a terapias nutricional e genética, o uso de suplementação com vitamina A e, recentemente, o uso do ácido valpróico, que é uma medicação usada, majoritariamente, em crises convulsivas, mas também utilizada em transtornos bipolares e profilaxia para enxaqueca, mas os resultados ainda carecem de estudos clínicos com maior número de pacientes, para que tenhamos deduções mais confiáveis e cientificamente válidas.
Aqueles pacientes que evoluem para uma baixa visual mais profunda e incapacitante podem e devem ser avaliados quanto ao uso de recursos ópticos que maximizem o residual de visão, numa das subespecialidades da Oftalmologia que é a Visão Subnormal.
Dadas às várias formas de transmissão genética desta doença, costumeiramente encaminhamos nossos pacientes a procurar uma avaliação com um geneticista, a fim de que sejam orientados quanto ao padrão genético de seu caso e os cuidados de uma eventual gravidez, para que possam estar cientes das chances de transmissão a sua prole.
Aguardamos para os próximos anos novidades principalmente aquelas relacionadas ao estudo com transplante de células tronco e o implante de chips na retina, o que certamente poderão ser divisores de água para esses pacientes e suas famílias.
